M-SADA is a method for assembling multiple conformational states of multidomain proteins through a population-based evolutionary algorithm, with multiple energy functions constructed by combining homologous and analogous templates with deep learning predicted inter-domain distance.
About M-SADA
What is M-SADA
How does M-SADA assemble protein domain structures to full-chain structure?
Starting from the input full-chain sequence and computationally predicted (or experimentally solved) domain structures, the full-chain structural analogues are detected from multidomain protein structure database (MPDB) according to the input protein domain models. Then, the homologous templates are successively searched by jackHMMER from MPDB, PDB, AlphaFold DB90 and AlphaFold DB according to the input full-chain sequence, where AlphaFold DB90 represents the models with an average (per-residue local distance difference test) pLDDT ≥90 in AlphaFold DB. Subsequently, the analogous and homologous templates are used to design multiple energy functions, which include different template restraints, physical constraints and residue distance information predicted by the in-house inter-domain distance prediction method DeepIDDP . A multiple population-based evolutionary algorithm is designed to explore and exploit the domain orientations based on multiple energy functions. Finally, full-chain models from different populations are selected and ranked using our developed model quality assessment method DeepUMQA2 .

Figure 1. Pipeline of M-SADA for multidomain protein multiple conformational state structures assembly.
What is the input of the M-SADA server?
Mandatory:
(1) A full-chain sequence in the standard FASTA format.
(2) At least 2 domain models in PDB format.
Optional:
Email address for receiving the results.
Name of the submitted job.
Sequence identity cutoff, for removing templates >the sequence identity cutoff with input full-chain sequence.
Please take note!!!
The following are some common problems:
(1) About input sequence:
The correct input form for a full-chain sequence:
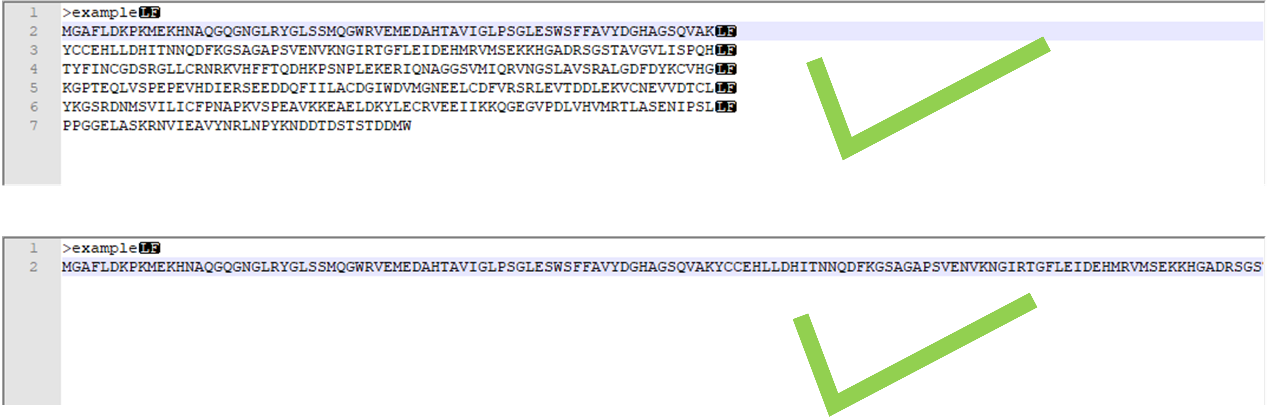
Figure 2. The correct input form for a full-chain sequence.
Common forms of error input sequence:
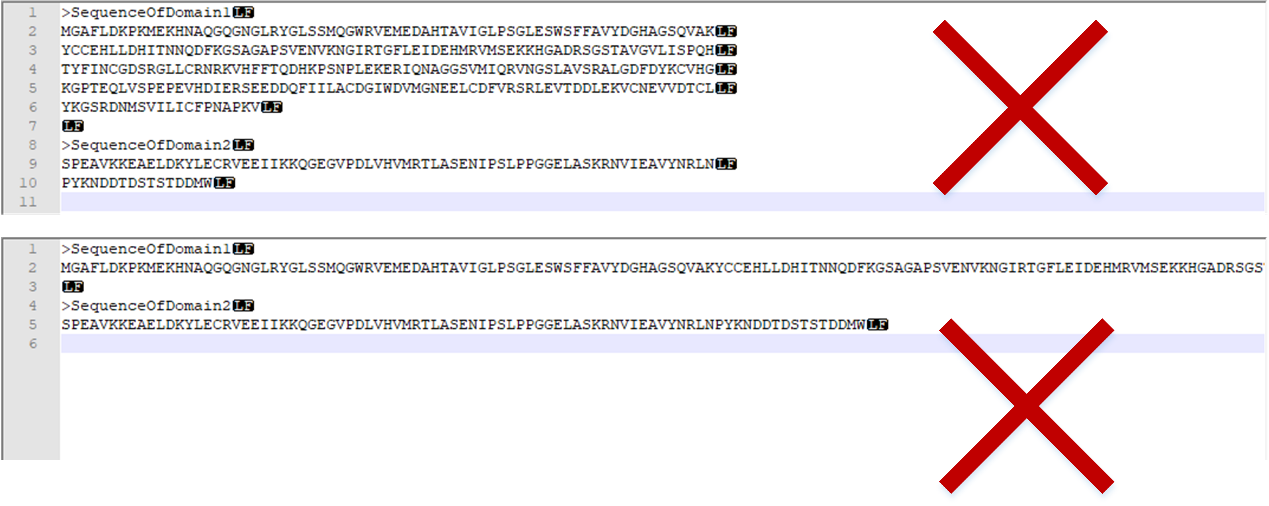
Figure 3. The incorrect input form for sequence.
(2) About input domain structures::
a.The residues in the input domain structures must correspond to the residues in the input sequence one by one. If there are missing residues in the domain structure (as shown in Figure 4), the program will not run properly.
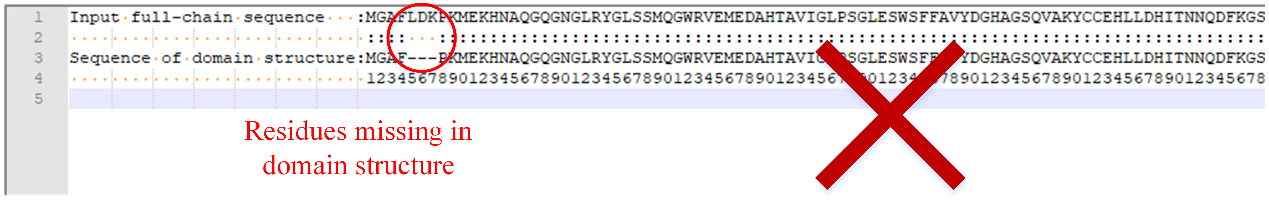
Figure 4. Residues missing in domain structure.
b.Each residue cannot contain more than one CA atom in the input protein domain structure (as shown in Figure 5). If a residue contains more than one CA atom, please remove the excess CA atoms. Otherwise, the program will not work properly.
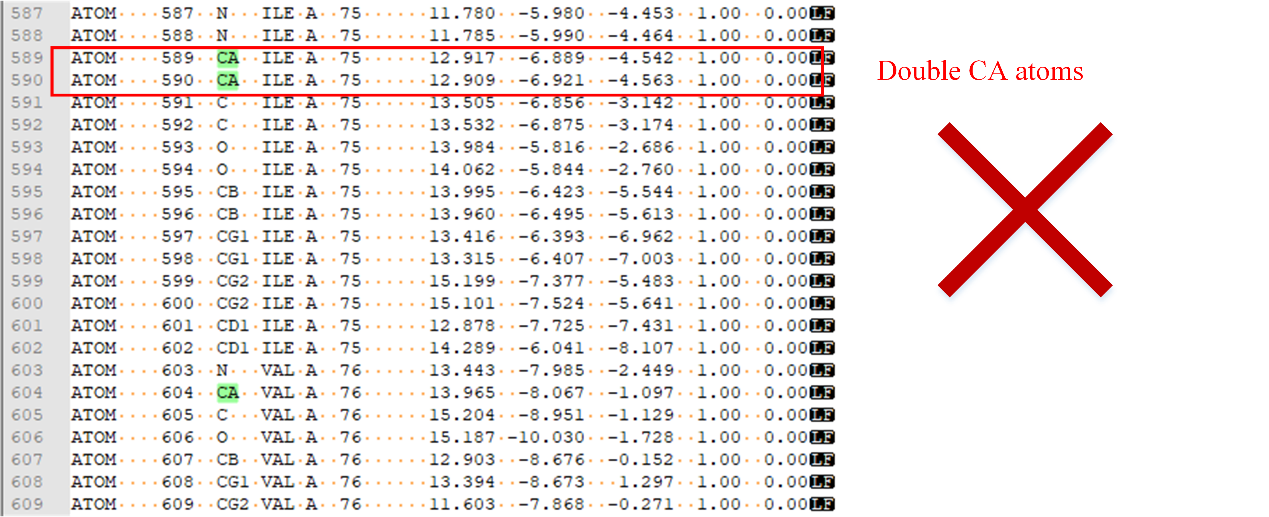
Figure 5. Double CA atoms.
How to run M-SADA?
Step1: upload full-chain sequence file (see Figure 6). Or input full-chain sequence information in text box (see Figure 7).
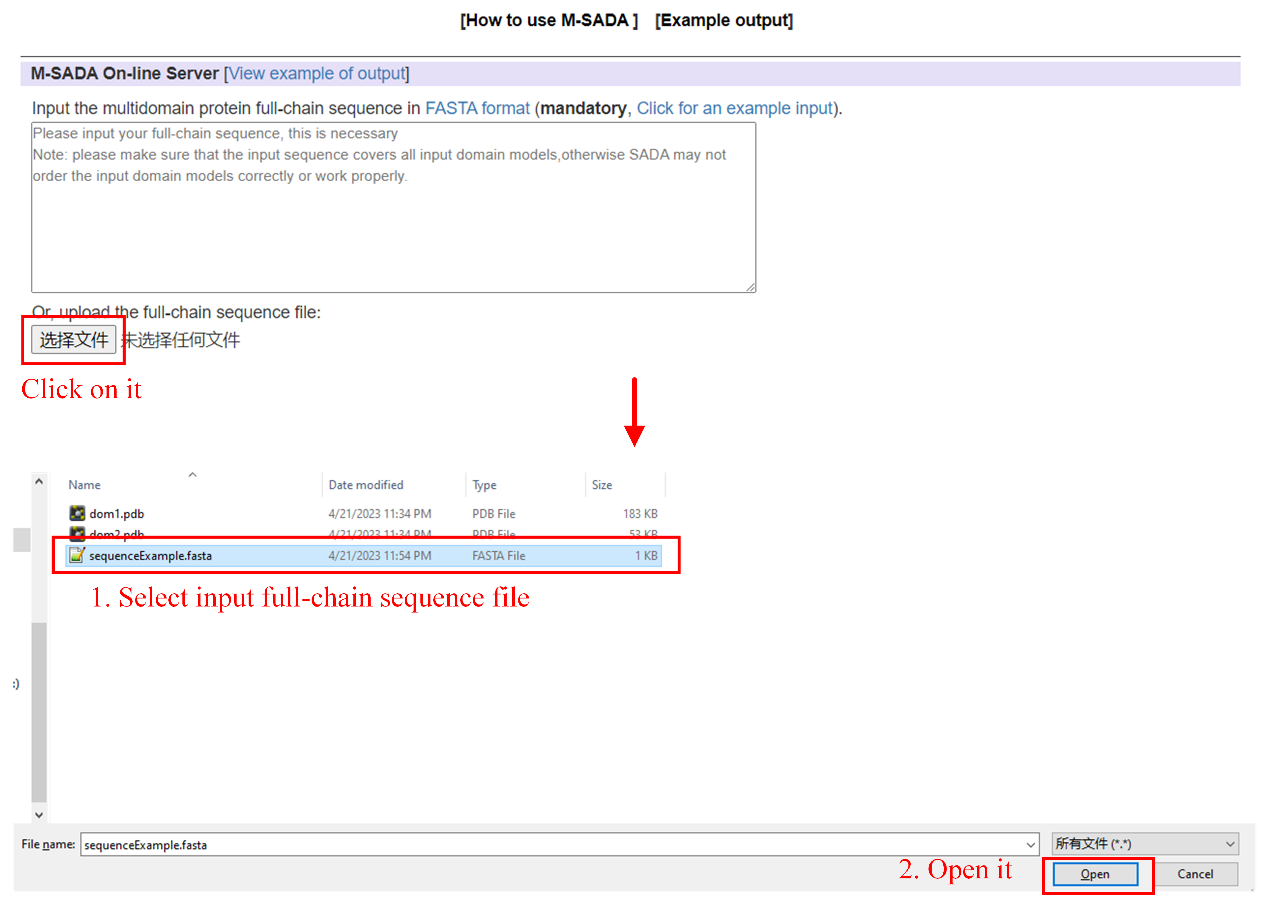
Figure 6. Upload sequence file.

Figure 7. Input full-chain sequence in the text box.
Step2: upload domain structure file (see Figure 8). Or input domain structure in text box (see Figure 9)
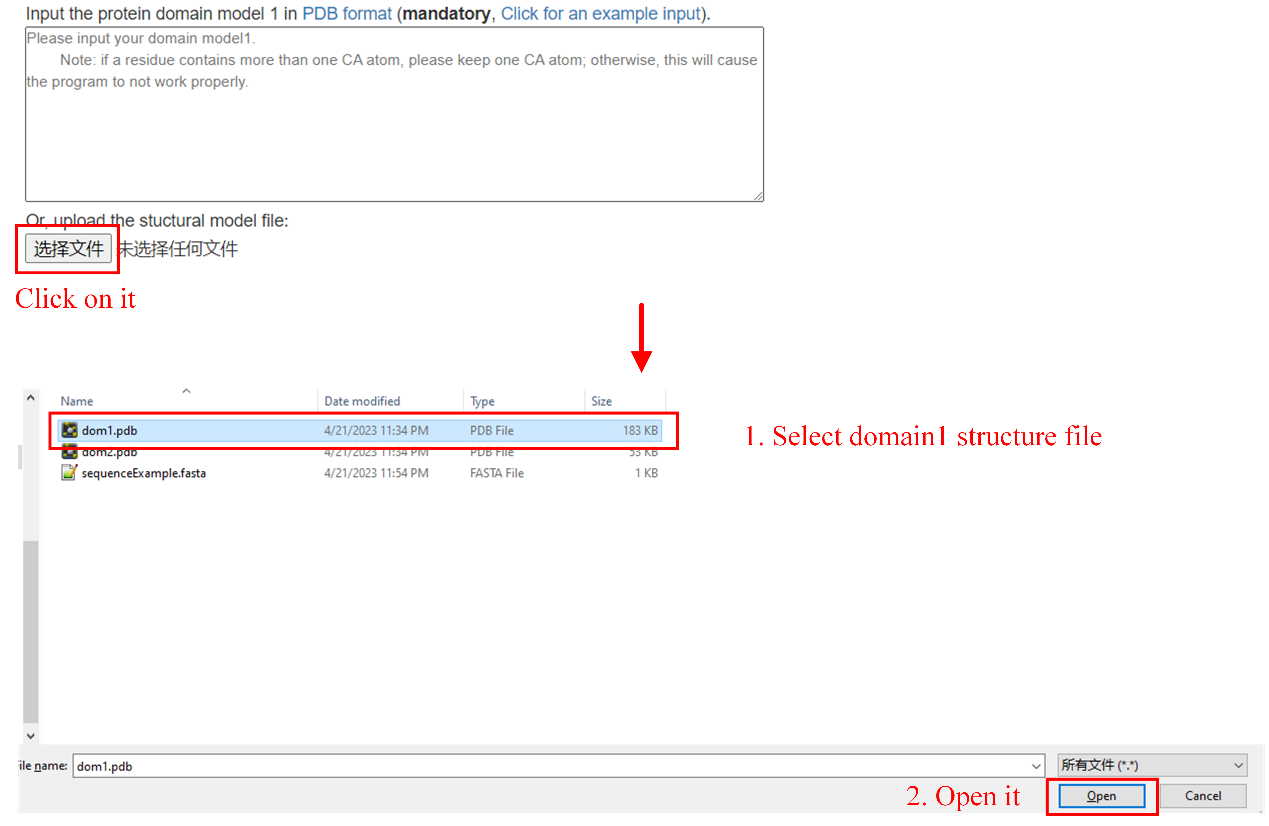
Figure 8. Upload domain structure file.

Figure 9. Input domain structure in text box.
Step3: Input the sequence identity cutoff (default 1.0), email (optional) and job name (optional), as see Figure 10.

Figure 10. Input job information.
Step4: Click on submit button to submit job.

Figure 11. Submit job.
Output of M-SADA server
(1) The full-chain models assembled
(2) Residue distance deviation of the full-chain structure model
(3) Per-residue lDDT and global lDDT of the full-chain structure model
(4) Ranking of all assembled full-chain structure models (according to the global lDDT)
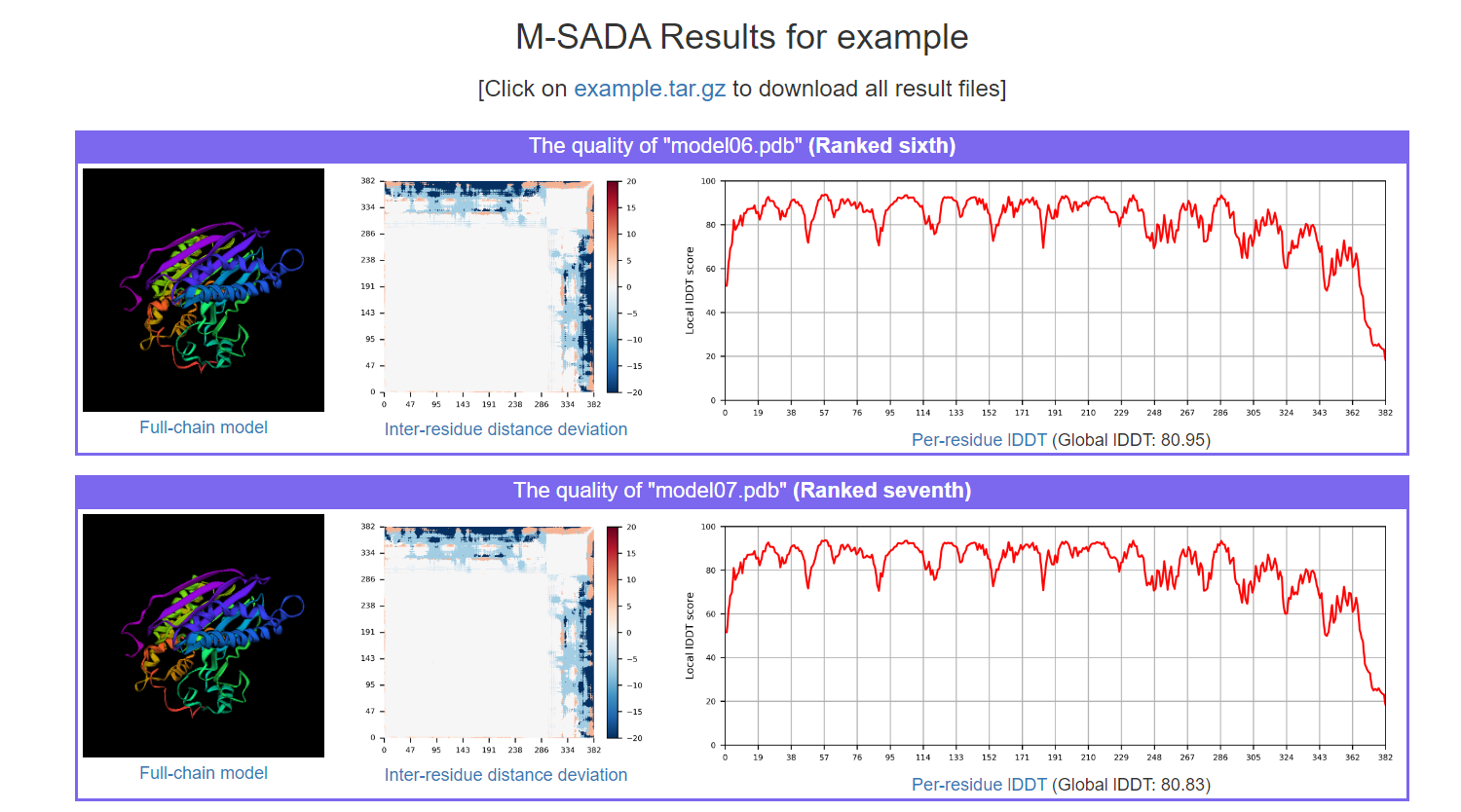
Figure 12. Output page of M-SADA.
Contact information
The M-SADA server is in active development with the goal to provide the most accurate multidomain protein structure assembly. Please help us achieve the goal by sending your questions, feedback, and comments to guijunzhanglab@163.com.